










Що таке фенілкетонурія (ФКУ)
Методичні рекомендації з діагностики і лікування ФКУ люб'язно надані фахівцями Краківського дитячого госпіталю
Фенілкетонурія (ФКУ) – це вроджена генетично обумовлене порушення метаболізму незамінної амінокислоти фенілаланіну (ФА), яка надходить в організм з харчовим білком, в тирозин (ТР). У нашій країні частота цього захворювання невелика: хвора дитина припадає на п'ять-шість тисяч здорових новонароджених. Причиною захворювання є мутація в гені гідроксилази фенілаланіну (ген ФАГ). В організмі хворої дитини відбувається накопичення надмірної кількості фенілаланіну в периферичній крові, а отже, і в ценральной нервовій системі (ЦНС). Надлишок фенілаланіну надає важкий токсичну дію на організм дитини. При відсутності своєчасної ранньої діагностики це невідворотно веде до важкої розумової та фізичної недорозвитку, будучи причиною ранньої дитячої інвалідності.
Згідно з даними Міністерства охорони здоров'я України за 2000 рік, утримання одного хворого з фенілкетонурією, починаючи від періоду новонародженості до 15 року життя при повному дієтичному лікуванні, досягає суми 150 000 доларів США. Витрати государсва на утримання дитини-інваліда без урахування витрат на скриниговые програми і дієтичне лікування складають близько 11 000 гривень. Завдяки ефективному скринінгу, а також своєчасному впровадженню лікування в більшості випадків стає можливим попередження появи неврологічних і психіатричних порушень, що в результаті сприяє нормальному фізичному і психічному розвитку хворих з ФКУ, а в майбутньому народженням у них здорового потомства.
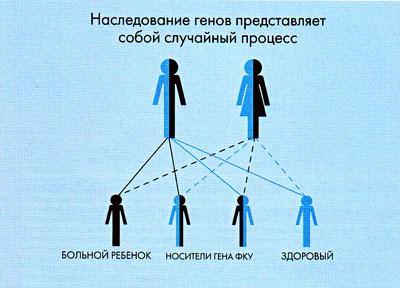 Хвора дитина з фенілкетонурією може народитися тільки в тій родині, де обоє батьків є носіями гена ФКУ. Визначити носійство гена ФКУ у батьківської пари можливо при генетичному обстеженні на гетерозиготное носійство, проведене в медико-генетичних центрах. Якщо в сім'ї вже є хвора дитина з ФКУ, носійство гена ФКУ у батьків очевидно. Однак навіть якщо обидва батьки є носіями гена ФКУ, їх діти не обов'язково хворі. Якщо прийняти за 100% всіх дітей, які гіпотетично можуть народитися в даній сім'ї, можна говорити про таке ризик виникнення захворювання ФКУ:
Хвора дитина з фенілкетонурією може народитися тільки в тій родині, де обоє батьків є носіями гена ФКУ. Визначити носійство гена ФКУ у батьківської пари можливо при генетичному обстеженні на гетерозиготное носійство, проведене в медико-генетичних центрах. Якщо в сім'ї вже є хвора дитина з ФКУ, носійство гена ФКУ у батьків очевидно. Однак навіть якщо обидва батьки є носіями гена ФКУ, їх діти не обов'язково хворі. Якщо прийняти за 100% всіх дітей, які гіпотетично можуть народитися в даній сім'ї, можна говорити про таке ризик виникнення захворювання ФКУ:
- ризик народження хворих дітей з ФКУ становить 25%;
- ризик народження дітей, які, подібно їхнім батькам, носіями гена ФКУ становить 50%;
- у решти 25% випадків народяться здорові діти
Відомо, що частота носійства мутантного гена ФКУ серед населення України становить 2 -3%. Частота захворювання 1:7000.
Дитина з ФКУ народжується без будь-яких проявів захворювання. Однак з початком годування, при надходженні в організм білка грудного молока або його замінників виникають перші симптоми, важко розпізнавані не тільки батьками, але і педіатрами.
Так, в періоді новонародженості до початку лікування у дитини з ФКУ можливі такі ознаки захворювання: необґрунтована млявість або неспокій; звертають на себе увагу розсіяний, блукаючий погляд, відсутність посмішки, слабке рухове пожвавлення. До 6 місяців у нього виявляється затримка психомоторного розвитку: він перестає активно реагувати на події; втрачає здатність впізнавати матір; не перевертається на живіт; не намагається сісти.
У другому півріччі життя батьки вже не можуть не помітити нерозуміння мови дорослого, невміння виражати голосом і мімікою дитини свої переживання. У дітей старше трьох років наростають розумова відсталість, збудливість, підвищена стомлюваність; порушується поведінка, що проявляється в розгальмуванні, психотичних розладах.
Часто у нелікованих хворих на фенілкетонурію сеча має своєрідний «мишачий» запах. Іноді виникають судомні напади різного ступеня вираженості; екзематозні зміни на шкірі.
Діагностика фенілкетонурії
 З численних спадкових захворювань обміну речовин (а їх налічується не менше 700) фенілкетонурія - найбільш «сприятливий», оскільки при ранній діагностиці можлива повна реабілітація хворого і його повноцінна адаптація до соціального життя, чого не можна досягти при багатьох інших видах спадкової патології. До сьогоднішнього дня питання ранньої діагностики у новонароджених захворювання ФКУ вирішене. Протягом останніх років відшліфовувалася методика масового обстеження (скринінг всіх новонароджених в нашій країні по виявленню захворювання на фенілкетонурію. Наказом Міністерства охорони здоров'я організовано забезпечення масового скринінгу новонароджених на фенілкетонурію на всій території України.
З численних спадкових захворювань обміну речовин (а їх налічується не менше 700) фенілкетонурія - найбільш «сприятливий», оскільки при ранній діагностиці можлива повна реабілітація хворого і його повноцінна адаптація до соціального життя, чого не можна досягти при багатьох інших видах спадкової патології. До сьогоднішнього дня питання ранньої діагностики у новонароджених захворювання ФКУ вирішене. Протягом останніх років відшліфовувалася методика масового обстеження (скринінг всіх новонароджених в нашій країні по виявленню захворювання на фенілкетонурію. Наказом Міністерства охорони здоров'я організовано забезпечення масового скринінгу новонароджених на фенілкетонурію на всій території України.
Скринінг-тест проводиться не раніше ніж на 3 доби (72 години) після народження дитини. При виявленні у дитини підвищеного рівня фенілаланіну (гиперфенилаланемии) скриниговая лабораторія погоджує тактику стосовно кожного пацієнта з провідним лікарем-генетиком.
При рівні ФА в скриниге:
- 3-8мг% - батьків або опікунів інформують про необхідність виконання контрольного дослідження (колориметричним методом). Якщо в котрольном дослідженні рівень ФА вище 2мг%, то необхідно сповістити батьків/опікунів і запросити в письмовій формі в спеціалізований діагностичний лікувальний центр;
- вище 8мг% - необхідно сповістити батьків/опікунів про результаті скринигового тіста і запросити в письмовій формі в спеціалізований діагностичний лікувальний центр.
Під час першого візиту лікар-спеціаліст (найчастіше педіатр або генетик) зобов'язаний надати батькам/опікунам вичерпну інформацію щодо передбачуваного захворювання, його причин і можливостей лікування.
У зв'язку з існуванням ряду причин появи гиперфенилаланемии (фенілкетонурія, нетипові форми фенілкетонурії і тирозенемия), необхідно провести диференціальну діагностику. Якщо підтвердиться дефіцит гідроксилази фенілаланіну (фенілкетонурія), застосовується дієтотерапія. У разі, якщо причиною виявиться порушення обміну речовин іншого роду, будуть обрані інші методи лікування.
Типи фенілкетонурії
Класична фенілкетонурія –phe > 1200 μмол/л (20 мг%)
Помірна фенілкетонурія – phe – 900 -1200 μмол/л (15 - 20 мг%)
М'яка фенілкетонурія – phe –600–900 μмол/л (10-15мг%)
М'яка гиперфенилаланинемия (gray zone) - phe – 360 – 600 μмол/л (6 - 10мг%)
М'яка гиперфенилаланинемия- не потребує лікування – phe – 120 – 360 μмол/л (2 - 6мг%)
(NIH PKU Conference report: State of the science and future research needs. Feb.22-23.2012)
Злоякісна гиперфенилаланинемия – недостатність тетрагідробіоптеріна (BH4)
Дефіцит гідроксилази фенілаланіну (PAH)
- фенілаланін ≤ 7 мг% (6 мг%) не потребує лікування – спостереження!
- фенілаланін > 7 мг% (6 мг%) – низкофенилаланиновая дієта
Дефіцит BH4 (злокачесивенная ФКУ) - фармакологічне лікування.
Клінічні появи злоякісної фенілкетонурії:
- важкі і швидко прогресуючі неврологічні порушення (несмотрия на нормальний рівень ФА в крові вследстве дієтотерапії)
- гіпотонія
- спастичний синдром
- атаксія
- порушення ковтання
- неспокій
- прогресуюча розумова відсталість
- важко купірується напади судом – найбільш характерні (до 3 міс. життя ознаки захворювання можуть бути відсутні)
- рівні фенілаланіну нехарактерні ( високі – як у класичній ФКУ або низькі - як у м'якій гиперфенилаланинемии).
Прогресуюче ураження нервової системи при злоякісній фенілкетонурії може вести до смерті дитенка, якщо захворювання не буде виявлено і не буде застосовано відповідне лікування.
Також про диференційованої діагностики читайте у статті Рекомендації з лікування фенілкетонурії в Польщі
Лікування фенілкетонурії
Лікування фенілкетонурії полягає в обмеженні надходження фенілаланіну з природними продуктами в харчуванні дитини. Це дозволяє попередити настання тяжких порушень ЦНС і забезпечити нормальний інтелектуальний розвиток. Показанням до початку дієти з низьким вмістом фенілаланіну є визначення концентрації ФА в сироватці крові >7-8мг% (згідно з рекомендаціями польських і німецьких центрів). Деякі центри (англійські та Європейської Федерації) рекомендують почати лікування при концентрації фенілаланіну >6,6 мг%.
Єдиним ефективним методом лікування хворих на ФКУ є спеціалізована дієтотерапія з моменту встановлення діагнозу.
Дієта при фенілкетонурії - це:
- Зменшення дози фенілаланіну відповідно до індивідуальної толерантності до фенилаланину, що означає зменшення дози натурального білка в добовому раціоні
- Забезпечення відповідної для нормального розвитку дози білка (додатковий білок без фенілаланіну) з продуктів лікувального харчування ФКУ
- Забезпечення відповідної дози енергії з використанням спеціальних продуктів низкобелковых
- Забезпечення відповідної дози вітамін, макро - і мікроелементів – головним чином препаратів ФКУ та інших джерел.
Амінокислотні суміші
 У пацієнтів із ФКУ кількість споживаного білка з натуральних продуктів не може перевищувати встановленої норми. У зв'язку з цим у маленьких дітей і у старших переважна частина потреби в білку, тобто близько 80%, має бути погашено сумішами, що не містять фенілаланін, збагаченими мінеральними інгредієнтами.
У пацієнтів із ФКУ кількість споживаного білка з натуральних продуктів не може перевищувати встановленої норми. У зв'язку з цим у маленьких дітей і у старших переважна частина потреби в білку, тобто близько 80%, має бути погашено сумішами, що не містять фенілаланін, збагаченими мінеральними інгредієнтами.
Добове споживання фенілаланіну з харчових продуктів повинно бути обмежено до такої кількості, щоб контрольований рівень концентрації фенілаланіну в сироватці крові не перевищував "безпечного для ЦНС" рівня, тобто 2-4мг/дл, це і є індивідуальна добова толерантність фенілаланіну. З метою повного задоволення потреб дитини з фенілкетонурією, і підтримання на допустимому рівні, що вживається з продуктів натурального білка і фенілаланіну слід всі харчові продукти відміряти і зважувати, а також вибирати продукти з низьким вмістом фенілаланіну.
Лікувальні амінокислотні суміші та препарати для хворих на ФКУ
Докладніше про методику розрахунку і корекції дієти читайте в розділі Дієта.
Спеціальна дієта з низьким вмістом фенілаланіну зазвичай запобігає несприятливі ефекти, що супроводжують це захворювання. Чим довше дитина перебуває на дієті, тим краще його інтелектуальний розвиток. При неадекватному розширенні або скасування дієти в підлітковому віці негативну дію фенілаланіну на організм не припиняється. Інтоксикація може призвести до зниження здатності до навчання, поведінкових проблем, аж до виникнення психічних розладів. Тому в деяких містах України, Росії та закордоном прийнято дотримання дієти до 18-річного віку. Останні європейські та американські спостереження говорять про те, що у випадку класичної і деяких інших форм ФКУ бажано довічне дотримання дієти.
Особливо важливо знати, що дівчата, які вступають у дітородний вік, повинні дотримуватися дієти до і під час вагітності. При недотриманні жінкою з ФКУ специфічної дієти під час вагітності у дитини виникають такі вади розвитку:
- у 92% випадків - розумова відсталість
- у 73% випадків - мікроцефалія
- у 12% випадків - вроджені вади серця
- у 40% випадків - низька маса тіла при народженні
Якщо жінка з фенілкетонурією як мінімум за 2 місяці до вагітності і під час вагітності дотримується дієти і рівень ФА не ревышает 4 мг%, дитина народиться здоровим.
Читайте також: Як жити з фенілкетонурією